This vignette demonstrates how examples in the SoS workflow manuscript (Wang G. and Peng B. 2019) are reproduced.
Requirement
To run the examples, SoS workflow system have to be installed. See here for an instruction. The interactive analysis part of the RNA-seq differential expression example additionally requires one to have a Jupyter environment (Jupyter Lab or Jupyter Notebook) installed, with R kernel. Although some examples are written in *.ipynb the Jupyter format, there is no need to install notebook related software to execute those notebooks from command line (as will be shown below).
If you edit plain *.sos file with vim editor you should see customized syntax highlighting being applied to *.sos script. To use SoS Notebook as IDE to edit examples in *.ipynb format, the SoS Notebook package has to be installed. Notice that the SoS workflow system only uses SoS Notebook as the IDE and does not rely on its cross-language data communication features. So there is no need to configure SoS Notebook for different kernels if your goal is to edit and run SoS workflow system.
SoS IDE
To demonstrate SoS IDE we prepared two versions of large scale regression example:
- Workflows in Jupyter format
*.ipynbfound on SoS live server.- Relevant examples are under
examples/WP_2019_SoS_Manuscript/WP2019_*.ipynb
- Relevant examples are under
- Workflows in plain text
*.sosfound in this repository.
Jupyter-based IDE
The Jupyter version can be executed directly on our live server. Jupyter-based IDE facilicates ad hoc analysis of workflow intermediate results, inspecting the outcome and making prototype extensions of workflows.
If you want to try out the large scale regression example without installing SoS, you can run them on our live server directly. At the end of each examples/WP2019_*.ipynb file there is a cell
%sosrun
Simply execute that cell using shift-enter -- you should have activated the workflows and get results. You can use
!ls
in a new cell to list contents in the folder and examine the results.
These examples are written in plain text format SoS script. Here is a screen-shot from vim editor:

Both *.ipynb and *.sos can be executed from command line via sos run. Here we demonstrate running SoS workflow from command line with *.sos scripts.
To extract narratives and possible command options from an SoS script, eg, from plain text script Process_Oriented.sos,
Or, for workflows in notebooks,
"Process oriented" style example: feature selection in machine learning
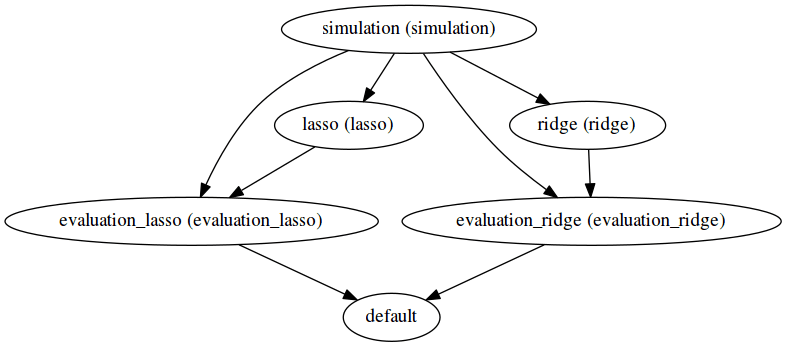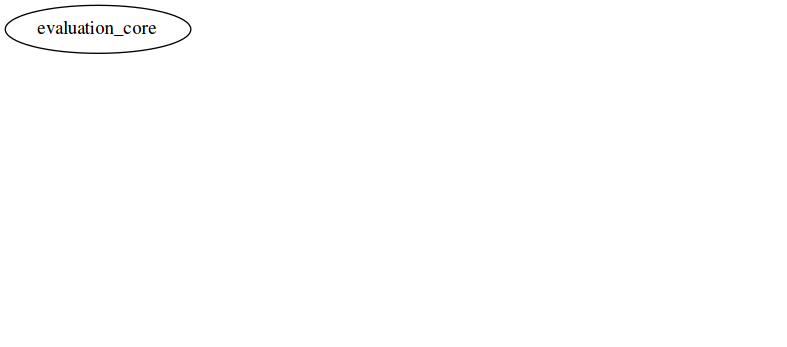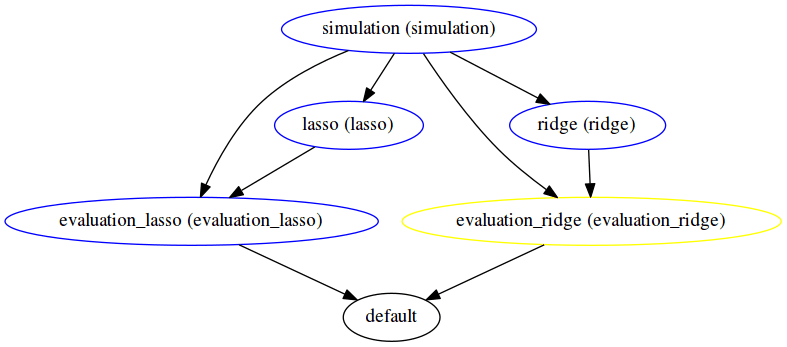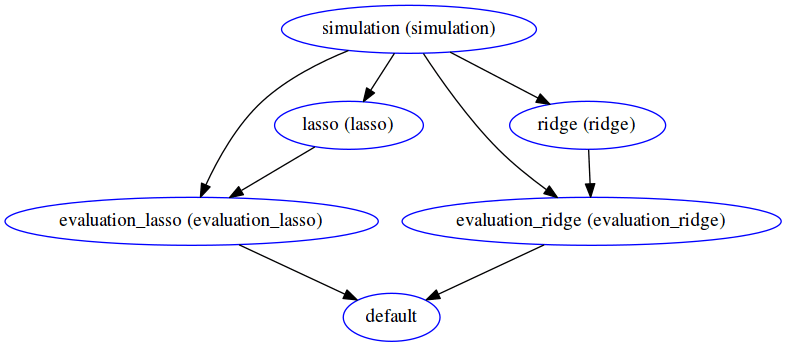
sos run executes the SoS file *.sos. Here we demonstrate additional options to report the execution status of each step (-p) and produce the DAG to summarize execution logic (-d).
In order for these options to work, Python packages graphviz, pillow and imageio are required. They can be installed via pip install, eg, pip install graphviz pillow imageio. Additionally you need to ensure dot executable is available. Oh Debian based Linux it can be installed via
apt-get install graphviz
Of course you can choose not to bother with these dependencies by dropping the -p and -d options. They will not effect the execution of the workflow.
Notice that each workflow step processes in parallel 5 replicates, generating 5 or 10 output files concurrently. This is what it is meant by "groups" in the output message above.
Package dependencies for this workflow
R and Python
The workflow requires R packages MASS and glmnet, and Python packages sklearn. If you do not have these packages on your system SoS will quit on error for relevant steps.
There are many ways to install these packages in R and Python. But caution that in Python to install sklearn via pip you need to type in scikit-learn instead, eg:
pip install scikit-learn==0.20.0
pandoc for generating reports
pandoc (version 2+) is needed to generate the HTML report.
The outcome of this workflow is a table of performance comparison, summarized in report.html. Here is the content of it:
Also available, due to -p and -d options, is a workflow status report automatically generated by the workflow. See here for the status report.
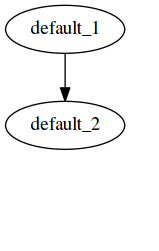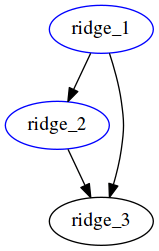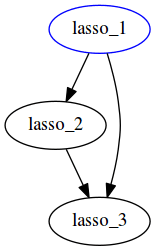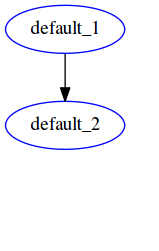
This workflow results in the same output as the previous one, in report.html:
The execution graph of this style can be found in the status report generated here. Notice the difference in the DAG generated -- here all file targets are incorporated into one DAG, compared to having 2 DAGs for the previous workflow for Lasso and ridge separately.

The output will be the same as previous styles, but execution graph is logically different from previous graphs, as shown in the status report here.

The execution logic of outcome oriented style with step targets is the same as that of outcome oriented style with file targets. But as one can tell from the difference in DAG, the dependencies are made more explicit and the DAG representation is more abstract yet clearer.
This example features the use of named_output to define dependencies for the process-oriented subworkflows. Because the dependency pattern is a lot less complicated than in Outcome Oriented style example, it can be sufficiently replaced by using a less powerful yet more intuitive implementation with named_output function.
Modular implementation of feature selection in machine learning
Examples above implemented several stand-alone *.sos scripts that contains all codes required to run the feature selection task and comparisons. Alternatively, one can created separate scripts (in R or Python in this example) to implement the core computation, and execute them in SoS. Here we give examples of such modular implementation of some pipelines above.
The module files are available under folder regression_modules. For example, the content of lasso.py is displayed below:
Pipelines Process_Oriented_Modular.sos and Outcome_Oriented_Modular.sos both use these modules. To run an example,
Whole-genome sequencing genotype calling pipeline
View the complete notebook here.
This pipeline implements a samtools tutorial of WGS calling. The major feature of SoS implementation is that one can summarize and preview intermediate results (see link above for details where we showed preview for a BAM file and a diagnostic plot). For this pipeline all steps are executed using pre-configured containers. Users do not have to deal with WGS genotyping software installations. However, docker is required to run this workflow. The installation process has in fact been documented in the WGS genotyping workflow. To reiterate here:
Docker installation on Linux
- Run commands below:
curl -fsSL get.docker.com -o get-docker.sh
sudo sh get-docker.sh
sudo usermod -aG docker $USER
- Log out and log back in
- Type
docker run hello-worldto verify installation
We have provided a toy data-set to run this pipeline. All steps in the pipeline are executed by docker. It will take a while to download them at some steps, if it is your first time running this pipeline.
View the complete notebook here.
The RNA-seq alignment pipeline is part of the RNA-seq differential analysis procedure that provides input data for the interactive DE analysis tutorial in Bioconductor. Using SoS Notebook or JupyterLab as IDE, we consolidate workflows with interactive analysis in one file. Here is a screenshot from the IDE:

The IDE comes with SoS syntax highlighting, and allows cell-by-cell execution of workflows. This notebook can also be executed from command terminal. The DE tutorial has many sections with codes, text and figures. When organized in a notebook file, a table of content can be displayed in the side panel to allow users to navigate between sections and edit relevant scripts. This helps with book-keeping of pieces of scattered code.
To checkout what workflows are offered:
Notice: this example involves running STAR RNA-seq aligner, which requires at least 32GB memory. Please run this workflow on a machine that has more than 32GB memory. In fact we have implemented in the workflow a check for memory resource and quit on error if the requirement is not satisfied:
system_resource(mem='40G', disk='200G')
To execute the alignment workflow on a computer with memory >40G, for one sample as a test:
Or, to align for all default samples:
It will take a while to prepare the human reference genome, download the data and complete the analysis. In the end, the RNA-seq data will be aligned to BAM format ready for interactive analysis in R.
The RNASeqGTEx.ipynb workflow example demonstrates executing computational steps as "external tasks" in separated computing environments. Notice the use of task statement for some steps, along with configurations. Two separate computing systems are used: the lab_server for big memory job, and uchicago_cluster for a number of CPU intensive computing. The jobs are submitted to remote computers but the results are kept in sync among machines. Due to restricted data access and computing environment requirement we will not demonstrate the execution in action, but users should refer to the Remote Execution documentation page on how to configure their system.
Here are two examples where SoS workflow has been used as companion pipelines to methods / software development projects,
- Data preprocessing for TADA-A R analysis, for Liu et al 2018, American Journal of Human Genetics.
- Data preprocessing for MASH R analysis, for Urbut et al 2018, Nature Genetics.
- Genetic fine-mapping applications, for methods discussed in Wang et al 2018 on biorxiv